
Behind the Numbers: A Conversation with Dr. Mark J. Sarno

Dr. Mark J. Sarno has over 40 years’ experience in research, development, clinical/regulatory, biostatistics, and project management within the pharmaceutical, in vitro diagnostics, and medical device fields. He has developed products covering such diverse clinical areas as:
fetal aneuploidies
newborn screening
thrombophilia
asthma
gastroenterology
oncology
cardiovascular disease
diabetes
infectious disease
metabolic bone disease
rheumatology
He is a patented inventor, member of the United States Patent Bar and an associate member of the California Lawyers Association. He founded Vision Biotechnology Consulting and Vision Clinical Research, which has assisted companies for 30 years in design, development, clinical research, and regulatory affairs resulting in 51 FDA clearances and approvals.
As a biostatistician, Dr. Sarno has analyzed some of the seminal epidemiological studies in cardiovascular disease including the Atherosclerosis Risk in Communities (ARIC) study and international oncology studies such as REDUCE. He has authored book chapters including a chapter in the Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, and published in many prestigious journals such as:
Clinical Chemistry
Clinical Biochemistry
Prostate Cancer and Prostatic Disease
Gynecologic Oncology
Fertility and Sterility
and Cancer Research
Journal of Urology
Urology
Diabetes Care
Food and Drug Law
Journal of the American College of Cardiology
Journal of Clinical Pharmacology
In his occasional spare time, Dr. Sarno enjoys running, flying airplanes, riding motorcycles, rescuing German Shepherds, and playing drums, keyboards, and bass guitar.





-
After spending a few years in academia, I actually started my industrial career as a product development scientist at an in vitro diagnostic (IVD) company. The company was quite progressive in their commitment to continuous education of their scientists. In my case, the company provided training courses in biostatistics, taught by Doug Montgomery, PhD, a statistician of great renown. Together with a few other scientists, we applied the training to product development, such as the use of factorial design and response surface modeling, as well as to clinical statistics. I found the knowledge and application of statistics to development efforts tremendously useful as it greatly improved our efficiency by quickly identifying factors contributing to product performance and to optimizing those factors. This profoundly helped us to achieve product design specifications and to assure product robustness. Beyond that, we could design, power, and analyze our own early clinical trials internally rather than working through third parties, and this provided a very rapid assessment of clinical validity. I would later apply similar concepts in pharmaceutical study design and analysis, including pharmacokinetic evaluations. In essence, the use of statistics greatly broadened and extended my perspectives on the translation of basic research to clinical application in the diagnosis, mitigation, and treatment of human disease.
-
We have worked extensively with risk factor biomarkers in the medical device field. Many of these target prognostic indications. As such, survival analysis provides a very powerful statistical methodology for performance assessment.
As an example, I worked on a prognostic for prediction of prostate cancer recurrence following radical prostatectomy. The test developer initially assessed early pilot studies purely by cross-sectional univariable parameters such as sensitivity and specificity for correlation with an outcome of biochemical recurrence. However, it was clear to me that regulatory bodies would view the marker as prognostic and that such a prognostic should correlate with a true clinical outcome such as death due to cancer progression. Further, the dimension of time to death should form part of the analysis. Thus, we pursued survival methods including, as a primary analysis, multivariable Cox Proportional Hazards regression adjusting for existing standard of care predictors and powered the study based on that method. The Food and Drug Administration (FDA) confirmed this approach as preferential to the cross-sectional parameters for correlation to biochemical recurrence. The test performed brilliantly in the trial with very high adjusted hazard ratios and was cleared for marketing.
-
To my mind, there isn’t much difference. Every study should acquire data with accuracy and integrity, data management should closely inspect the data prior to lock, and the analyses should follow the pre-specified Statistical Analysis Plan (SAP).
I suppose the only difference that comes to mind relates to the magnitude of captured data. As we know, pharmaceutical studies may acquire a large body of data from each study subject with numerous variables arising from physical examinations, laboratory testing, imaging, and other measures, and that powered Phase III studies may enroll thousands of subjects followed over a great period of time. As such, data inspection, analysis, and quality control may require a greater period of time and effort. It is vital that programming in SAS (or other suitable platform) should occur well ahead of time with close QC of the programming so that generation of tables, figures, and listings can occur within a suitable timeframe following data lock.
-
I would say that both human and software resources drive rigor and integrity. Certainly, it starts with development and approval of a robust clinical protocol and associated SAP taking into account capture of all necessary primary, secondary, and exploratory variables. From there, a robust electronic data capture (EDC) system must be designed and validated, thus requiring excellent and experienced EDC programmers and a suitable EDC platform. Then, clinical monitors must ensure thorough clinical site training and data integrity via frequent remote and on-site monitoring and timely and clear query generation and resolution. Lastly, data managers must maintain close contact and inspection of data with programmed frequent backups. All these human and electronic efforts are conditioned proportionately upon the degree of complexity of the study with higher complexity requiring increased dedication of human efforts and more sophisticated software platforms. In contrast, novel technologies don’t necessarily require greater rigor or integrity unless the captured variables themselves are novel. In such a case, early discussion with regulatory bodies should help identify any additional efforts beyond current industry best practices.
-
The most important aspect of collaboration to my mind is clear communication of the desired Intended Use for the product from the development team, which should include personnel from R&D, regulatory, clinical, quality, marketing, and manufacturing. Once the organization specifies the desired Intended Use, the statistician can then begin to think about:
study design,
primary analysis statistical method,
and powering.
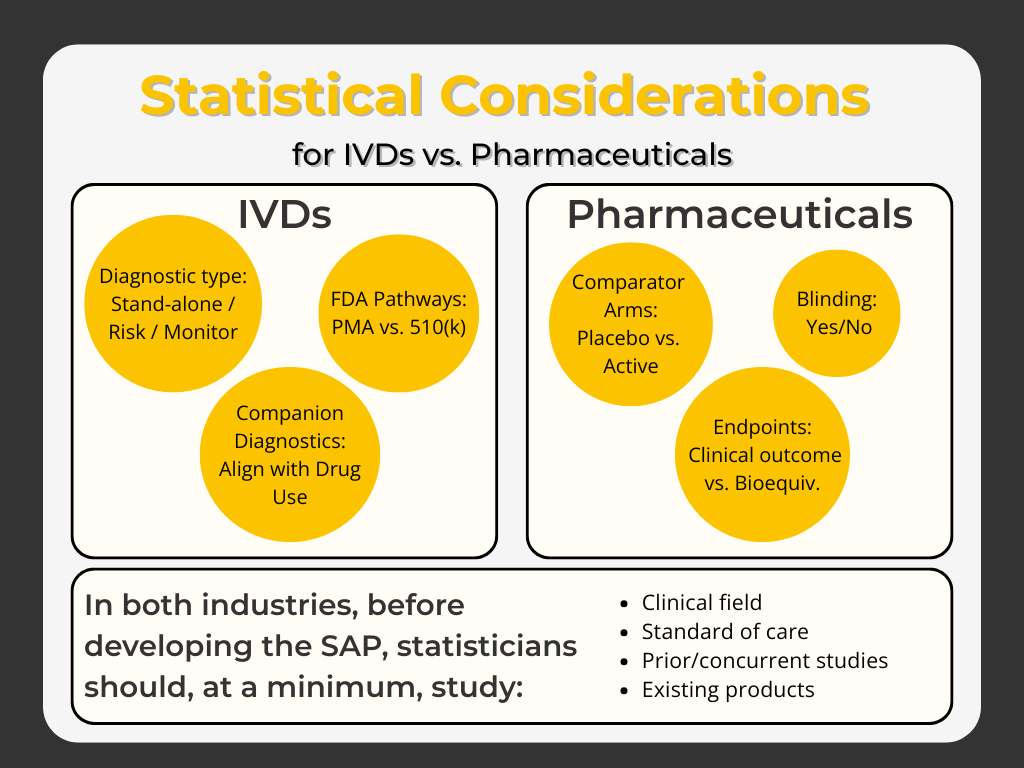
In the case of IVDs, the statistician must know if the test shall embody a stand-alone diagnostic, risk factor, or monitoring test since these indications will require very different study designs and statistical methods and will be reviewed by the FDA under different pathways (Pre-Market Approval [PMA] vs. Section 510(k)).
Extending this discussion, a companion diagnostic for a drug will require even greater attention to Intended Use, how it shall be used with the drug, and the regulatory expectations. For pharmaceuticals, similar questions arise such as comparator arms (placebo versus active reference drug), blinding, clinical outcome versus demonstration of bioequivalence under Section 505(b)(2), etc.
For both industries, the statistician should make a careful study of the clinical field, the current standard of care, prior and contemporaneous research and regulatory studies, and existing products prior to development of the SAP.
-
The unfortunate truth is that, for an independent or CRO statistician, it is very difficult to keep up with the many guidelines applicable to the broad variety of clinical fields in which one may work. Rather, I find the best approach is to search the FDA (and/or EMA) system for applicable guidelines when each new project presents itself. In some cases, the search may not turn up anything new and thus the statistician relies on the existing guidelines of which they are already aware. But, when new guidances appear, I take time to absorb them in detail and consider applicability to the project. In those few cases where I have had questions, I have contacted the guidance author directly and they have generally responded quickly.
-
I don’t generally find any communication issues with regulatory bodies, as the review team almost always has a Biometrics team member available. Further, many clinicians, particularly those with Masters of Public Health (MPH) degrees, have solid backgrounds in statistics and can quickly relate to the concepts and results under discussion. One possible exception is that many clinicians relate to parameters such as sensitivity, specificity, and positive and negative predictive values but find it hard to grasp the meaning and impact of parameters like hazard ratios from survival analysis or odds ratios from logistic regression. In contrast, communicating these and other more esoteric concepts (e.g. multiple imputation techniques, propensity scores, etc.) can induce more confusion than clarity when presenting to marketing teams or other non-scientist stakeholders.
The only way to reduce the confusion is to avoid technical jargon as much as possible and present the methodology in simple terms.
As an example, when attempting to explain a hazard ratio of 1.6, I might say that this value represents, in the case of a pharmaceutical, a 60% greater risk of encountering a clinical event in the absence of taking the drug studied. By the way, we would typically take the inverse of the hazard ratio in order to present the risk reduction by taking the drug but this can confuse non-statisticians as well.
Suffice it to say, it is not possible to avoid some confusion in these situations but the statistician must do the best they can.
-
In my experience, this has happened quite rarely. If the sponsor develops a robust product, crafts an appropriate Intended Use, and enters into early discussions with regulatory bodies as to appropriate studies and expected performance, unexpected challenges should not occur.
The only instance I can recall was in a pharmacokinetic study. In those studies, the standard approach is to utilize model-independent/non-compartmental analysis. However, in one case, I felt the data in some patients reflected a 2-compartment distribution and so I performed model-dependent/compartmental analysis in an exploratory fashion to assess the presence of a second compartment. This was not pre-specified in the SAP but I deemed it prudent to investigate. In the end, the indications were not sufficient to conclude a second compartment, but at least we had performed the analysis to exclude the possibility.
-
It is a given that you will be strong in math and you will embrace thoroughly the statistical methods you learn. But, it is also of vital importance that you take time with each new project to learn at least the basics of the clinical field. No one expects you to become a luminary clinician in the field; but you should know the recognized signs, symptoms, diagnosis, and treatment paradigms of the disease in order to properly apply statistics to study design, powering, and analysis of efficacy studies for new products.
-
At first blush, I would point to healthcare economic modeling. It’s not new but I think it needs more work. I say that because I believe both false negative and false positive results have occurred in this field with potentially beneficial medical devices or drugs apparently not demonstrating positive healthcare benefits and possibly non-beneficial products apparently demonstrating healthcare benefits.
Basically, I believe these types of studies require more attention, more sound design and rigor than they have received to date. In fact, I feel as though there are only a few centers of excellence in this regard yet almost every medical device and drug institution is required to do these studies.
We need to develop consensus methods to these studies and this may require attention in education of biostatisticians, application in academia, and guidances in industry. Those biostatisticians who become expert in this field may find themselves in high demand.

Mark J. Sarno, eJD
Chief Executive Officer | Biostatistician | Project Director | Legal Officer
Vision Clinical Research, LLC
760-815-4086